ADN
Problema a resolver
El ADN, el portador de la información genética en los seres vivos, se ha utilizado en la justicia penal durante décadas. ¿Pero cómo funciona exactamente la elaboración de perfiles de ADN? Dada una secuencia de ADN, ¿cómo pueden los investigadores forenses identificar a quién pertenece?
En un archivo denominado dna.py en una carpeta denominada dna, implemente un programa que identifique a quién pertenece una secuencia de ADN.
Demostración
Código de distribución
Para este problema, ampliarás la funcionalidad del código que te proporcionó el personal de CS50.
Inicia sesión en cs50.dev, haz clic en la ventana de terminal y ejecuta cd por sí solo. Deberás ver que el mensaje de la ventana de terminal se asemeja al siguiente:
$
Luego ejecuta
wget https://cdn.cs50.net/2023/fall/psets/6/dna.zip
para descargar un ZIP llamado dna.zip en tu espacio de códigos.
Luego ejecuta
unzip dna.zip
para crear una carpeta llamada dna. Ya no necesitas el archivo ZIP, por lo que puedes ejecutar
rm dna.zip
y responder con "y" seguido de Enter en el indicador para eliminar el archivo ZIP que descargaste.
Ahora escribe
cd dna
seguido de Enter para moverte (es decir, abrir) ese directorio. El mensaje ahora debería parecerse al siguiente.
dna/ $
Ejecuta ls solo y deberías ver algunos archivos y carpetas:
databases/ dna.py sequences/
Si tienes algún problema, ¡sigue estos mismos pasos de nuevo y observa si puedes determinar dónde te equivocaste!
Antecedentes
El ADN es en realidad solo una secuencia de moléculas llamadas nucleótidos, dispuestas en una forma particular (una doble hélice). Cada célula humana tiene miles de millones de nucleótidos dispuestos en secuencia. Cada nucleótido de ADN contiene una de cuatro bases diferentes: adenina (A), citosina (C), guanina (G) o timina (T). Algunas partes de esta secuencia (es decir, genoma) son las mismas, o al menos muy similares, en casi todos los humanos, pero otras partes de la secuencia tienen una mayor diversidad genética y, por lo tanto, varían más en la población.
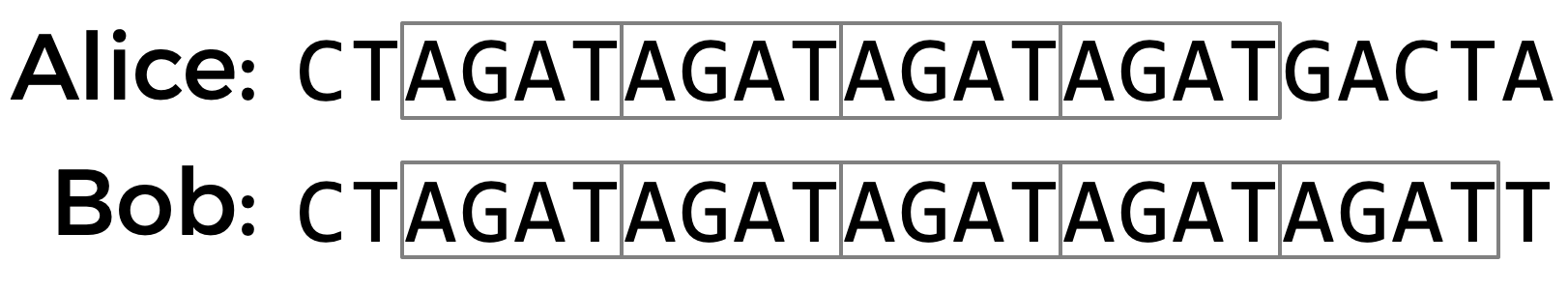
Un lugar donde el ADN tiende a tener una gran diversidad genética es en las repeticiones cortas en tándem (STR). Una STR es una secuencia corta de bases de ADN que tiende a repetirse consecutivamente numerosas veces en ubicaciones específicas dentro del ADN de una persona. El número de veces que se repite cualquier STR en particular varía mucho entre individuos. En las muestras de ADN a continuación, por ejemplo, Alice tiene la STR AGAT repetida cuatro veces en su ADN, mientras que Bob tiene la misma STR repetida cinco veces.
El uso de múltiples STR, en lugar de uno solo, puede mejorar la precisión de la elaboración de perfiles de ADN. Si la probabilidad de que dos personas tengan el mismo número de repeticiones para una sola STR es del 5% y el analista observa 10 STR diferentes, entonces la probabilidad de que dos muestras de ADN coincidan puramente por casualidad es aproximadamente de 1 entre un billón (suponiendo que todas las STR sean independientes entre sí). Entonces, si dos muestras de ADN coinciden en el número de repeticiones para cada una de las STR, el analista puede estar bastante seguro de que provienen de la misma persona. CODIS, la base de datos de ADN del FBI, utiliza 20 STR diferentes como parte de su proceso de elaboración de perfiles de ADN.
¿Cómo podría verse una base de datos de ADN de este tipo? Bueno, en su forma más simple, podrías imaginar formatear una base de datos de ADN como un archivo CSV, en el que cada fila corresponde a un individuo y cada columna corresponde a una STR particular.
nombre,AGAT,AATG,TATC
Alicia,28,42,14
Bob,17,22,19
Charlie,36,18,25
Los datos en el archivo anterior sugerirían que Alice tiene la secuencia AGAT repetida 28 veces consecutivas en algún lugar de su ADN, la secuencia AATG repetida 42 veces y TATC repetida 14 veces. Bob, mientras tanto, tiene esas mismas tres STR repetidas 17 veces, 22 veces y 19 veces, respectivamente. Y Charlie tiene esas mismas tres STR repetidas 36, 18 y 25 veces, respectivamente.
Entonces, dada una secuencia de ADN, ¿cómo podrías identificar a quién pertenece? Bueno, imagina que miras a través de la secuencia de ADN la secuencia consecutiva más larga de AGAT repetidos y descubres que la secuencia más larga tenía 17 repeticiones. Si luego encuentras que la secuencia más larga de AATG es de 22 repeticiones y la secuencia más larga de TATC es de 19 repeticiones, eso proporcionaría una buena evidencia de que el ADN era de Bob. Por supuesto, también es posible que una vez que tomes los conteos de cada una de las STR, no coincida con nadie en tu base de datos de ADN, en cuyo caso no tienes coincidencia.
En la práctica, dado que los analistas saben en qué cromosoma y en qué ubicación del ADN se encontrará una STR, pueden localizar su búsqueda solo en una sección estrecha de ADN. Pero ignoraremos ese detalle por este problema.
Tu tarea es escribir un programa que tome una secuencia de ADN y un archivo CSV que contenga conteos de STR para una lista de individuos y luego imprima a quién pertenece el ADN (muy probablemente).
Especificación
- El programa debe requerir como su primer argumento de línea de comandos el nombre de un archivo CSV que contenga los conteos de STR para una lista de individuos y debe requerir como su segundo argumento de línea de comandos el nombre de un archivo de texto que contenga la secuencia de ADN para identificar.
- Si tu programa se ejecuta con un número incorrecto de argumentos de línea de comandos, tu programa debe imprimir un mensaje de error de tu elección (con
imprimir). Si se proporciona el número correcto de argumentos, puedes asumir que el primer argumento es de hecho el nombre de archivo de un archivo CSV válido y que el segundo argumento es el nombre de archivo de un archivo de texto válido.
- Si tu programa se ejecuta con un número incorrecto de argumentos de línea de comandos, tu programa debe imprimir un mensaje de error de tu elección (con
- Tu programa debe abrir el archivo CSV y leer su contenido en la memoria.
- Puedes suponer que la primera fila del archivo CSV serán los nombres de las columnas. La primera columna será la palabra
nombrey las columnas restantes serán las secuencias STR mismas.
- Puedes suponer que la primera fila del archivo CSV serán los nombres de las columnas. La primera columna será la palabra
- Tu programa debe abrir la secuencia de ADN y leer su contenido en la memoria.
- Para cada una de las STR (de la primera línea del archivo CSV), tu programa debe calcular la ejecución más larga de repeticiones consecutivas de la STR en la secuencia de ADN para identificar. ¡Ten en cuenta que hemos definido una función auxiliar para ti,
longest_match, que hará precisamente eso! - Si los conteos de STR coinciden exactamente con cualquiera de los individuos en el archivo CSV, tu programa debe imprimir el nombre del individuo coincidente.
- Puedes suponer que los conteos de STR no coincidirán con más de un individuo.
- Si los conteos de STR no coinciden exactamente con ninguno de los individuos en el archivo CSV, tu programa debe imprimir
No coincide.
Consejos
-
Puede resultar útil el módulo
csvde Python para leer archivos CSV y guardarlos en la memoria. De particular ayuda podría sercsv.DictReader.-
Por ejemplo, si un archivo como
foo.csvtiene una fila de encabezado, en la cual cada cadena es el nombre de algún campo, aquí le mostramos cómo podría imprimir esosfieldnamescomo unalista: ```python import csvwith open("foo.csv") as file: reader = csv.DictReader(file) print(reader.fieldnames) ```
-
Y aquí le mostramos cómo leer todas las (otras) filas de un CSV en una
lista, en la cual cada elemento es undictque representa esa fila: ```python import csvrows = [] with open("foo.csv") as file: reader = csv.DictReader(file) for row in reader: rows.append(row) ```
-
-
Las funciones
openyreadtambién podrían resultar útiles para leer archivos de texto hacia la memoria. - Piense en las estructuras de datos que podrían ser útiles para mantener un seguimiento de la información en el programa. Una
listao undictpodrían resultar útiles. - Recuerde que definimos una función (
longest_match) que, dadas una secuencia de ADN y un STR como entradas, devuelve el número máximo de veces que se repite el STR. ¡Luego puede usar esa función en otras partes del programa!
Tutorial
Cómo realizar la prueba
Aunque check50 esté disponible para este problema, le recomendamos que primero pruebe su código por su cuenta para cada uno de los siguientes.
- Ejecute el programa como
python dna.py databases/small.csv sequences/1.txt. Su programa debería mostrarBob. - Ejecute el programa como
python dna.py databases/small.csv sequences/2.txt. Su programa debería mostrarNo match. - Ejecute el programa como
python dna.py databases/small.csv sequences/3.txt. Su programa debería mostrarNo match. - Ejecute el programa como
python dna.py databases/small.csv sequences/4.txt. Su programa debería mostrarAlice. - Ejecute el programa como
python dna.py databases/large.csv sequences/5.txt. Su programa debería mostrarLavender. - Ejecute el programa como
python dna.py databases/large.csv sequences/6.txt. Su programa debería mostrarLuna. - Ejecute el programa como
python dna.py databases/large.csv sequences/7.txt. Su programa debería mostrarRon. - Ejecute el programa como
python dna.py databases/large.csv sequences/8.txt. Su programa debería mostrarGinny. - Ejecute el programa como
python dna.py databases/large.csv sequences/9.txt. Su programa debería mostrarDraco. - Ejecute el programa como
python dna.py databases/large.csv sequences/10.txt. Su programa debería mostrarAlbus. - Ejecute el programa como
python dna.py databases/large.csv sequences/11.txt. Su programa debería mostrarHermione. - Ejecute el programa como
python dna.py databases/large.csv sequences/12.txt. Su programa debería mostrarLily. - Ejecute el programa como
python dna.py databases/large.csv sequences/13.txt. Su programa debería mostrarNo match. - Ejecute el programa como
python dna.py databases/large.csv sequences/14.txt. Su programa debería mostrarSeverus. - Ejecute el programa como
python dna.py databases/large.csv sequences/15.txt. Su programa debería mostrarSirius. - Ejecute el programa como
python dna.py databases/large.csv sequences/16.txt. Su programa debería mostrarNo match. - Ejecute el programa como
python dna.py databases/large.csv sequences/17.txt. Su programa debería mostrarHarry. - Ejecute el programa como
python dna.py databases/large.csv sequences/18.txt. Su programa debería mostrarNo match. - Ejecute el programa como
python dna.py databases/large.csv sequences/19.txt. Su programa debería mostrarFred. - Ejecute el programa como
python dna.py databases/large.csv sequences/20.txt. Su programa debería mostrarNo match.
Corrección
check50 cs50/problems/2024/x/dna
Estilo
style50 dna.py
Cómo enviar
submit50 cs50/problems/2024/x/dna